

Hebah Fatafta, Mohammed Khaled, Batuhan Kav, Olujide O. Olubiyi, Birgit Strodel
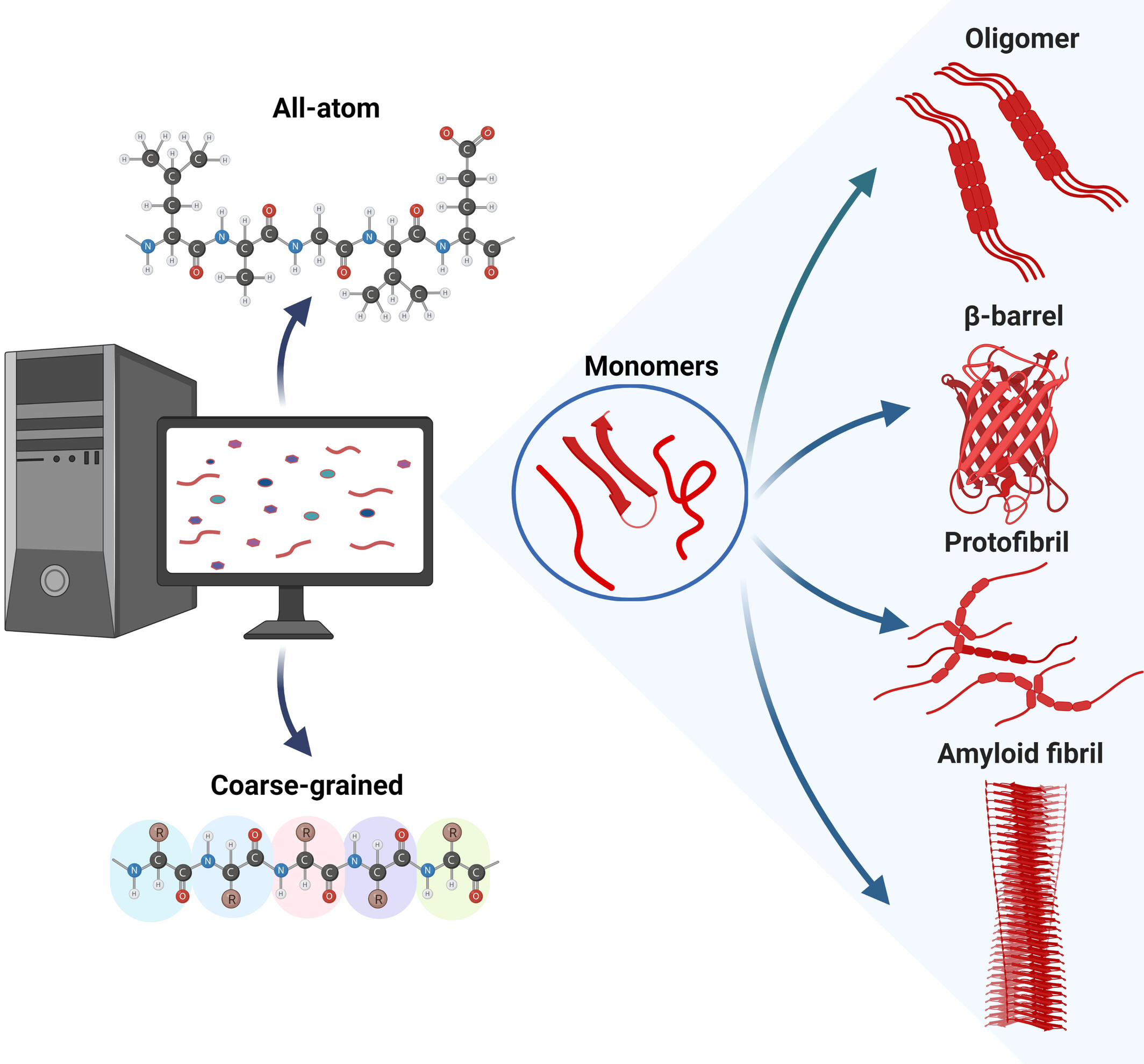
Amyloid proteins are characterized by their tendency to aggregate into amyloid fibrils, which are often associated with devastating diseases. Aggregation pathways typically involve unfolding or misfolding of monomeric proteins andformation of transient oligomers and protofibrils before the final aggregation product is formed. The conformational dynamics and polymorphic and volatile nature of these aggregation intermediates make their characterization by experimental techniques alone insufficient and also require computational approaches. Over the past 25 years, the size of simulated amyloid aggregation systems and the length of these simulations have increased significantly. These advances are discussed here. The review includes simulation approaches that model the aggregating peptides or proteins at both the all-atom and coarse-grained levels, use molecular dynamics simulations or Monte Carlo sampling to simulate the conformational changes, and present results for various amyloid peptides and proteins ranging from Lys-Phe-Phe-Glu (KFFE) as the smallest system to Aβ as an intermediate-sized peptide to α-synuclein. The presentation of the history of amyloid aggregation simulations concludes with a discussion of where the future of these simulations may lie.
https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1703